Abstract
Figure
Discussion
ReferencesAll material courtesy of LB 145 Team Bolt (Mike Moran and Nikki Verkest) |
The W1282X mutation of the Cystic Fibrosis gene results in a truncated CFTR protein and can lead to severe pulmonary disease and pancreatitis (Chang et al, 2007; Frulloni et al, 2002). The aim of this experiment was to create a reliable assay to detect the genetic disorder caused by this specific mutation, allowing early treatment procedures to begin to increase the quality of life for the affected individual and to increase the accuracy for prenatal and newborn screening procedures. Using our designed primers and protocol, the W1282X mutation was detected and the DNA sequence replicated with reduced error as calculated for the Taq Polymerase used. The primers were designed by identifying both the wild type and mutant DNA sequence around position 162604, along with a sequence 1200 basepairs away from the mutation and the complimentary sequences. Using PCR and gel electrophoresis, wild type and mutant DNA were readily identified using the developed primers and by viewing the amplified product in the agarose gel. The PCR ran through thirty cycles with an annealing temperature of 46 degrees C. Our assay detected the W1282X mutation within large DNA smears caused by the low annealing temperature used (Feriotto et al, 2001). Using a predicted error rate of 0.00525 errors/basepair (roughly 6.3 total errors for the replicated strands), we can safely assume that our replicated DNA sequences are in fact the sequences occurring within the DNA samples and that the W1282X locus sequence is preserved (Ling et al, 1991).
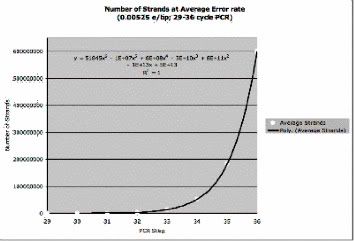 Refined error rate calculation of Taq Polymerase. Due to a high majority of PCR protocols requiring between thirty and thirty-five cycles, the data was further refined to include these cycles. While error is reduced for lower numbers of cycles, using too few cycles may reduce the visibility of the final products after gel electrophoresis. The six-degree polynomial fit generated allows a degree of estimation for the amount of product that has an average error and can be used for cycle numbers outside of this range, but the divergence increases drastically, especially for cycles greater than 36. Cystic Fibrosis, caused by a mutation on the CFTR gene, has many different forms and can be caused by many different variations of the mutation on the gene (Welsh and Smith, 1995). Mutation W1282X, which causes a truncation of the CFTR protein due to a Tryptophan codon being changed to a Stop codon, can lead to pancreatitis and pulmonary disease (Chang et al, 2007; Frulloni et al, 2002). Detecting this specific mutation within a given DNA sample is possible by performing PCR and analyzing the products using gel electrophoresis, as the amplified products will be 1200-bp lengths of replicated DNA when using both the wild-type and mutant primer pairs. Based on preliminary research into the replication error of Taq Polymerase and the W1282X mutation, we predicted that our designed primers and PCR protocol would reliably determine if a DNA sample has the mutation with negligible error. Our primers were designed to consist of twenty-two base pairs, allowing specific annealing to the correct DNA sequence and reliable amplification of that sequence. While primers of shorter lengths have been shown to be as reliable, a longer length was chosen to diminish the possibility of non-specific binding (Feriotto et al, 2001). These primers are located roughly 1200 basepairs apart, with the reverse primer situated on Intron 19 and the mutant and control primers situated on Exon 20. This relatively short replication length aided in minimizing the replication of the Taq Polymerase, as fewer basepairs are replicated using the enzyme (Ling et al, 1991). The primer location for the mutant and control primers was chosen to encompass the W1282X mutation (at Position 162604 on the DNA sequence) so that this mutation is preserved in the replicated products. The PCR protocol was chosen to have thirty cycles within each reaction in order to further minimize this replication error, as a greater number of cycles will increase the amount of error propagated through the reaction products. Our successful gels and error rate analysis show a combination of these error reduction tactics in our replicated products. While the band sizes differ from the expected length, this discrepancy can be assigned to come from the 1 kb MW ladder analysis. The exponential fits, while close to having a negligible uncertainty, over-estimate the band size length around the 1200-basepair length, as the fit line exists above the same line connecting the 1636- and 1016-basepair band lengths in both regressions. It can then be viewed that the viewed bands exist near the 1200-basepair mark and are our replicated products. These replicated products are predicted to have an average error of 6.3 basepairs, i.e. we expect that roughly six basepairs have been mutated during the PCR process from the original sequence based on the imperfections of Taq Polymerase (Ling et al, 1991; Keohavong et al, 1989). This error, however, only occurs between the two primers and do not affect the W1282X mutation locus. Our replicated strands then have a 99.5% match with the template DNA, as 6.3 of the 1200 basepairs have changed. The error rate, however, is only the average within the entire reaction products. By selecting a random strand to compare to the template nucleotide sequence, it is entirely possible to select one of the original strands of DNA or one with double the errors. This is due to the fact that, once a strand is replicated, it gains errors due to the polymerase. The original strand is then not affected, so in assuming that the template DNA had no errors (aside from the W1282X mutation or any targeted mutation), there will be some strands within the final reaction cocktail that display no errors when compared to the template DNA sequence (Bracho et al, 1998). This error was determined by calculating the number of strands within the PCR cocktail after each cycle at a certain error rate and averaging across the entire range of products. Based on the replication process, those error rates near the middle of the spectrum will collect more strands than those near the extremes, similar to a Gaussian distribution. We can, however, predict that any error created within the replicated strands do not affect the validity of stating that every product contains the W1282X mutation locus, whether mutated or not. As the Taq Polymerase only works between the primers, and the mutant and control primers were designed to anneal to the mutation site, the twenty-two basepair sequences of DNA are retained throughout the entire PCR process. The experimental procedure was also gradually refined through both our control research and the numerous replications of the W1282X analysis. By performing a high number of PCRs with the E. coli, Lambda virus, and the IB3 DNA, our team avoided many errors that could have hampered the validity of the assay. In the beginning stages of research, each post-electrophoresis gel showed primer bands at the bottom of the gel, indicating that the annealing temperature was too high since the primers could not anneal to the DNA. A total of 6 different PCR cocktails failed before finally getting results on the 7th through 10th PCR trials. The specific annealing temperatures and times were gradually refined until results were obtained, ensuring that the replicated DNA was indicative of the desired products. Care was taken to reduce the amount of non-specific binding caused by either too low of an annealing temperature or too long of an annealing time length, as this can lead to increased non-specific binding (Wittwer et al, 1993). We did, however, encounter some non-specific binding, but not enough that distinct bands within the gel could not be viewed; the non-specific binding occurred at seemingly random points along the initial DNA sequence, creating the DNA smears seen in most of the wells (Figures 4 and 6). The exact reaction cocktail volumes used in our final PCR protocols are also representative of the refinement done through the number of full experiments done. As slight differences in microliters can drastically alter the outcome of the PCR, either by making results indistinguishable from each other or preventing any results from being seen, our reactions cocktail volumes were the product of gradual refinement. The agarose gel creation and gel electrophoresis was also watched to ensure proper protocol adherence, and no further refinement was needed in these two areas. This experiment can be further refined if given more time. The low annealing temperature was due to the design of the reverse primer, leading to some of the non-specific binding viewed in various wells post-electrophoresis. This primer should be redesigned in order to narrow the gap between the annealing temperatures of the two forward primers (mutant and control) and the reverse primer. This will increase the annealing temperature used during PCR to a more-standard temperature, eliminating the need to reprogram a thermocycler between assays (Wittwer et al, 1993). A second reverse primer should also be designed to further differentiate between the mutant and wild-type DNA, as this second primer will be situated a different distance from the W1282X locus and create a different band length post-electrophoresis. The IB3 cells, and any DNA sample used to test the assay, will also be purified as near as possible to the time of running the PCR, as storing DNA samples for extended periods may degrade the sample (Cukier et al, 2009). By combining this PCR assay and other that also detect CFTR mutations, a multispectral assay could be developed to detect multiple mutations. This assay would then be able to detect a high percentage of mutations that cause Cystic Fibrosis, increasing the chance of detecting the disease in early stages. If genetic manipulation technology advances, the specific mutations may be able to be altered. This would lessen the severity of or eliminate Cystic Fibrosis in the human population. Bracho, M., A. Moya, and E. Barrio. 1998. Contribution of Taq polymerase-induced errors to the estmation of RNA virus diversity. Journal of General Virology. 79: 2921-2928. Chang, M., T. Chang, Y. Tien, P. Liang, I. Jan, N. Su, and J. Wong. 2007. Spectrum of mutations and variants/haplotypes of CFTR and genotype-phenotype correlation in idiopathic chronic pancreatitis and controls in Chinese by complete analysis. Clinical Genetics. 71: 530-539. Cukier, H., M. Pericak-Vance, J. Gilbert, and D. Hedges. 2009. Sample degradation leads to false-positive copy number variation calls in multiplex real-time polymerase chain reaction assays. Analytical Biochemistry. 386 (2): 288-290. Feriotto, G., A. Ferlini, A. Ravani, E. Calzolari, C. Mischiati, N. Bianchi, and R. Gambari. 2001. Biosensor Technology for Real-Time Detection of the Cystic Fibrosis W1282X Mutation in CFTR. Human Mutation. 18: 70-81. Frulloni, L., C. Castellani, P. Bovo, B. Vaona, B. Calore, G. Liani, G. Mastella, and G. Cavallini. 2002. Natural history of pancreatitis with cystic fibrosis gene mutations. Digestive and Liver Disease. 35: 179-185. Keohavong, P. and W. Thilly. 1989. Fidelity of DNA polymerases in DNA amplification. Proceedings of the National Academy of Science, USA. 86: 9253-9257. Ling, L., P. Keohavong, C. Dias, and W. Thilly. 1991. Optimization of the Polymerase Chain Reaction with regard to fidelity: Modified T7, Taq, and Vent DNA Polymerases. Genome Research. 1: 63-69. O'Keffe, H., X. Yao, J. Coull, M. Fuchs, and M. Egholm. 1996. Peptide nucleic acid pre-gel hybridization: An alternative to Southern hybridization. Proceedings of the National Academy of Science, USA. 93: 14670-14675. Welsh, M. and A. Smith. 1995. Cystic Fibrosis. Scientific American. 273 (6): 52-59. Wittwer, C., B. Marshall, B. Reed, and J. Cherry. 1993. Rapid Cycle Allele-Specific Amplification: Studies with the Cystic Fibrosis DeltaF-508 Locus. Clinical Chemistry. 39 (5): 804-809. |
|
Webpage © 2009 Team BOLT |